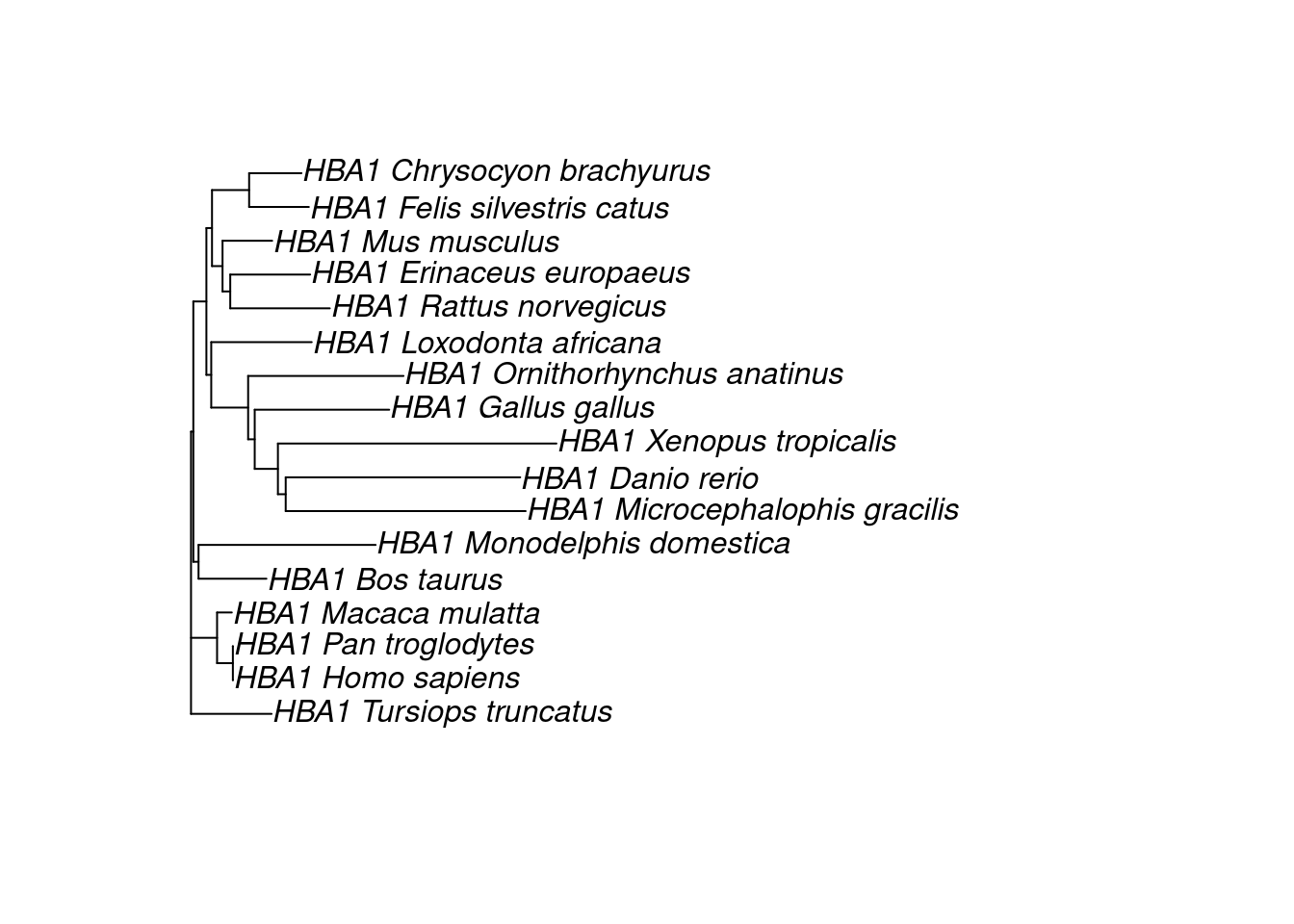
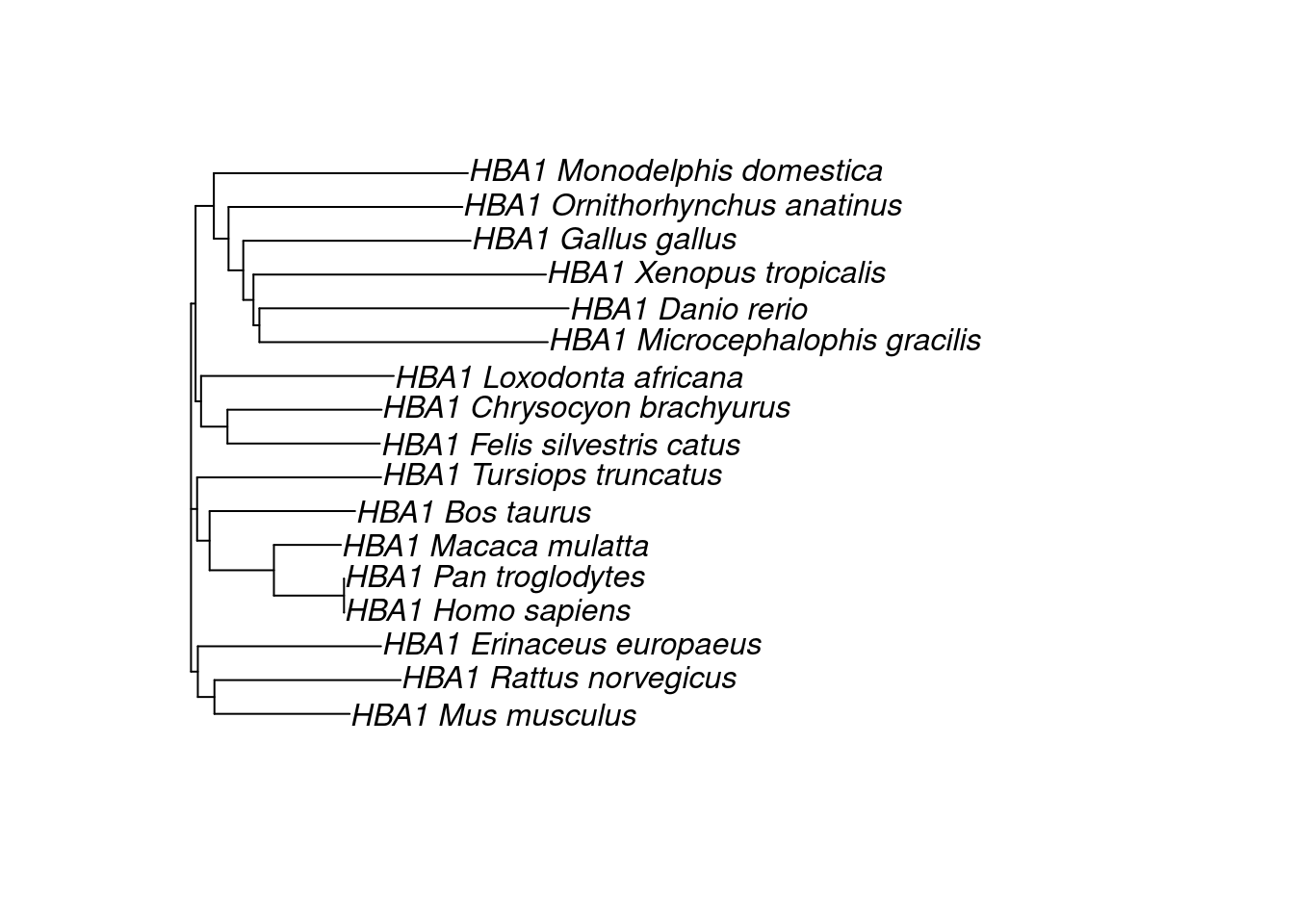
The simplest way to infer a phylogeny is to use pairwise distances between sequences, which are calculated from Multiple Sequence Alignments (MSA). Pairwise distances are typically stored in distance matrices, which are used to infer a phylogenetic tree using algorithms such as UPGMA or neighbor-joining. In this lesson, we will guide on you on how to infer a phylogeny from sequences.
3.1 Goals of this lesson
In this lesson, you will learn to:
perform MSA
calculate distances from MSA
infer phylogenetic trees from distance matrices
3.2 Multiple Sequence Alignment (MSA)
There are several algorithms to perform an MSA, such as ClustalW (Thompson, Higgins, and Gibson 1994), ClustalOmega (Sievers et al. 2011), MUSCLE (Edgar 2004), and MAFFT (Katoh and Standley 2013). Comparing them and deciding which one is the best is beyond the scope of this lesson. Here, we will perform MSA with the Bioconductor package msa (Bodenhofer et al. 2015), which provides native implementations of the ClustalW, ClustalOmega, and MUSCLE algorithms.
msa comes with an example FASTA file containing protein sequences of hemoglobin subunit alpha 1 in multiple species. Let’s start by reading the example file:
library(msa)library(Biostrings)# Path to example filefasta_path <-system.file("examples", "HemoglobinAA.fasta", package="msa")fasta_path
Now, we will use the function msa() to perform an MSA using the default algorithm (ClustalW). You can also use other algorithms by changing the argument passed to the method parameter.
Now that we have an MSA, we can calculate distances.
3.3 Calculating distance matrices
Here, we will calculate distance matrices using 2 different methods:
the aastring2dist() function from MSA2dist, which calculates distances from AAStringSet objects using a score matrix. Here, we will use the Grantham matrix (Grantham 1974).
the dist.alignment() function from seqinr, which calculates distances from alignment objects using a similarity or identity matrix. Here, we will use the identity matrix.
First, as these 2 functions take different data classes as input, we will have to convert the hemoglobin_aln object to the right classes for each function.
# Convert `MsaAAMultipleAlignment` to `AAStringSet` for MSA2distaln_msa2dist <-as(hemoglobin_aln, "AAStringSet")# Convert `MsaAAMultipleAlignment` to `alignment` for seqinraln_seqinr <-msaConvert(hemoglobin_aln, type ="seqinr::alignment")
The two methods lead to different tree topologies. How do you know which one is “true”, then? This is the topic of the next lesson, where you will learn how to use maximum likelihood to assess trees.
Exercises
Use the dist_matrix1 object to infer a phylogeny using the upgma() function from the phangorn package (Schliep 2011). Is the topology the same as the topology of tree1?
Solution
# Infer UPGMA tree and compare topologiestree_upgma <- phangorn::upgma(dist_matrix1)plot(tree_upgma)
References
Bodenhofer, Ulrich, Enrico Bonatesta, Christoph Horejš-Kainrath, and Sepp Hochreiter. 2015. “Msa: An r Package for Multiple Sequence Alignment.”Bioinformatics 31 (24): 3997–99.
Edgar, Robert C. 2004. “MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput.”Nucleic Acids Research 32 (5): 1792–97.
Grantham, Richard. 1974. “Amino Acid Difference Formula to Help Explain Protein Evolution.”Science 185 (4154): 862–64.
Katoh, Kazutaka, and Daron M Standley. 2013. “MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.”Molecular Biology and Evolution 30 (4): 772–80.
Sievers, Fabian, Andreas Wilm, David Dineen, Toby J Gibson, Kevin Karplus, Weizhong Li, Rodrigo Lopez, et al. 2011. “Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega.”Molecular Systems Biology 7 (1): 539.
Thompson, Julie D, Desmond G Higgins, and Toby J Gibson. 1994. “CLUSTAL w: Improving the Sensitivity of Progressive Multiple Sequence Alignment Through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice.”Nucleic Acids Research 22 (22): 4673–80.